بیماری آرپی (RP) چیست و چرا به آن مبتلا میشویم؟
بیماری رتینیت پیگمنتوزا (Retinitis Pigmentosa) یا به اختصار آرپی (RP)، شایعترین نوع از دیستروفیهای ارثی شبکیه (IRD) است که از هر ۳۰۰۰ تا ۵۰۰۰ نفر، یک نفر را درگیر میکند. شبکیه انسان دارای دو نوع سلول گیرنده نوری است: سلولهای استوانهای (Rods) که مسئول دید در تاریکی و دید محیطی هستند، و سلولهای مخروطی (Cones) که دید مرکزی، دقیق و رنگی را فراهم میکنند.
در آرپی، یک خطای ژنتیکی (جهش در یکی از بیش از ۱۰۰ ژن مرتبط مانند RHO یا USH2A) باعث میشود که سلولهای استوانهای دچار مرگ برنامهریزیشده شوند. با از بین رفتن آنها، سلولهای مخروطی نیز به تدریج آسیب میبینند.
الف) توارث اتوزومال غالب (Autosomal Dominant RP – adRP)
- میزان اپیدمیولوژی و شیوع نسبی: این الگوی توارث تقریباً ۲۰ الی ۳۰ درصد از کل جمعیت مبتلایان به رتینیت پیگمنتوزا غیرسندرمی را به خود اختصاص میدهد که نشاندهنده فراوانی نسبتاً بالای آن در مقایسه با الگوهای غیرمتعارف است.
- مکانیسم انتقال بیولوژیکی و ژنتیکی: انتقال فنوتیپ آسیبشناختی بر روی کروموزومهای غیرجنسی (اتوزوم) صورت میپذیرد و بر اساس اصول ژنتیک مندلی، حضور تنها یک آلل جهشیافته از سوی یکی از والدین جهت بروز کامل فنوتیپ بیماری در نسل بعد کفایت میکند. احتمال آماری انتقال آلل معیوب به هر یک از فرزندان، بدون وابستگی به جنسیت آنان، معادل ۵۰ درصد ارزیابی میگردد. از منظر زیستشناسی مولکولی، این پدیده عمدتاً ناشی از دو مکانیسم عمده است: «نارسایی هاپلوئیدی» (Haploinsufficiency) که در آن مقدار پروتئین سالم تولیدشده از تکآلل وحشی برای حفظ بقای سلولی کفایت نمیکند، و یا مکانیسم «غالب منفی» (Dominant-Negative effect) که در آن دگره جهشیافته محصولی غیرعملکردی تولید میکند که مانع فعالیت بیولوژیکی پروتئینهای هومولوگ طبیعی میگردد.
- سیر طبیعی و پیشآگهی کلینیکی: سیر بالینی و روند دژنراسیون سلولهای استوانهای در الگوی adRP در قیاس با سایر الگوها (بهویژه الگوی وابسته به جنس) بهطور معناداری ملایمتر و تدریجیتر گزارش شده است. اگرچه تظاهرات اولیه درگیری لایه فوتورسپتور ممکن است در دهههای دوم و سوم حیات بارز گردد، لکن بسیاری از مبتلایان دید مرکزی و تواناییهای عملکردی بینایی خود را تا دهه پنجم یا ششم زندگی به شکل پایداری حفظ مینمایند. از ویژگیهای بارز کلینیکی در این فرم، پدیده «بیانپذیری متغیر» (Variable Expressivity) و «نفوذ ناقص» (Incomplete Penetrance) بهویژه در لوکوسهای خاص نظیر ژن PRPF31 است؛ این رخداد بیولوژیکی سبب میگردد که شدت تظاهرات فنوتیپی در اعضای یک خانواده با جهش ژنتیکی همسان، تنوع چشمگیری را نشان دهد و حتی برخی ناقلان ژن، تا انتهای حیات فاقد هرگونه تظاهرات پاتولوژیک بالینی باشند.
- ژنهای شاخص و پاتوژنز مولکولی: ژن RHO واقع بر روی کروموزوم 3q22.1 که کدکننده اپوپروتئین رودوپسین است، به عنوان متداولترین فاکتور پاتوژنیک مولکولی در این الگو شناخته میشود و مسئول بروز تقریباً ۲۵ درصد از موارد adRP و ۱۰ درصد از کل گستره بیماری آرپی است. جهشهای موضعی در این ژن منجر به تاخوردگی نادرست ساختاری (Misfolding) پروتئین رودوپسین، تجمع تودهای آن در شبکه آندوپلاسمی، القای استرس شدید سلولی (ER Stress) و نهایتاً آغاز آبشار مرگ برنامهریزیشده سلول (Apoptosis) در فوتورسپتورهای استوانهای میگردد. فراتر از ژن رودوپسین، جهش در ژنهای ناظر بر فاکتورهای پیرایش آرانآ (Splicing Factors) نظیر PRPF31، PRPF8 و SNRNP200 و همچنین ژنهای ساختاری مژکهای سلولی مانند RP1، سهم عمدهای در ایجاد و پیشبرد پاتولوژی مولکولی این الگوی وراثتی دارند.
ب) توارث اتوزومال مغلوب (Autosomal Recessive RP – arRP)
- میزان شیوع: حدود ۳۵ تا ۴۰ درصد موارد.
- نحوه انتقال: فرزند باید دو کپی از ژن معیوب را همزمان از پدر و مادر ناقل خود دریافت کند. این الگو به دلیل وفور ناقلان خاموش، در ازدواجهای فامیلی بهشدت شایع است.
- ژنهای شاخص: ژن USH2A شایعترین ژن شناساییشده در این دسته است که با سندرم آشر (همراهی آرپی با ناشنوایی حسی-عصبی) نیز پیوند مستقیم دارد.
ج) توارث وابسته به جنس مغلوب (X-Linked Recessive RP – xlRP)
- میزان شیوع: حدود ۱۰ تا ۱۵ درصد موارد.
- نحوه انتقال: ژن جهشیافته روی کروموزوم X قرار دارد. به همین دلیل، تقریباً فقط مردان به فرم بالینی بیماری مبتلا میشوند و زنان عموماً ناقلان بدون علامت یا با تظاهرات بسیار خفیف هستند.
- پیشآگهی بالینی: این الگو شدیدترین و سریعترین روند پیشرفت دژنراسیون را دارد و بیماران معمولاً تا دهه سوم یا چهارم زندگی دچار آسیبهای شدید ساختاری شبکیه میشوند.
- ژنهای شاخص: ژنهای RPGR و RP2 متولیان اصلی این نوع انتقال هستند.
به نقل از وبسایت آکادمی چشمپزشکی آمریکا (AAO)
متن انگلیسی:
“Genetic testing has transitionally evolved from an academic exercise to an essential clinical standard of care for retinitis pigmentosa. Identifying the exact causative gene mutation is critical not only to confirm the inheritance pattern but also to determine patient eligibility for FDA-approved gene therapies and active clinical trials.”
ترجمه فارسی:
«آزمایش ژنتیک از یک اقدام آکادمیک به یک استاندارد بالینی ضروری در مراقبت از بیماران مبتلا به رتینیت پیگمنتوزا تبدیل شده است. شناسایی دقیق جهش ژنی مسبب، نه تنها برای تایید الگوی وراثت حیاتی است، بلکه جهت تعیین کاندیداتوری بیمار برای دریافت ژندرمانیهای تایید شده توسط FDA و شرکت در کارآزماییهای بالینی فعال، نقشی کلیدی ایفا میکند.»
علائم بیماری آرپی چشم؛ از شبکوری تا دید تونلی
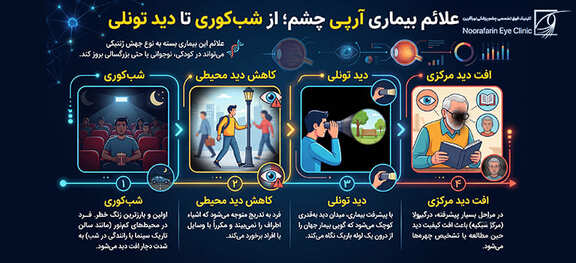
علائم این بیماری بسته به نوع جهش ژنتیکی میتواند در کودکی، نوجوانی یا حتی بزرگسالی بروز کند. مسیر معمول علائم به این شرح است:
- شبکوری (Nyctalopia): اولین و بارزترین زنگ خطر. فرد در محیطهای کمنور (مانند سالن تاریک سینما یا رانندگی در شب) به شدت دچار افت دید میشود.
- کاهش دید محیطی: فرد به تدریج متوجه میشود که اشیاء اطراف را نمیبیند و مکرراً با وسایل یا افراد برخورد میکند.
- دید تونلی (Tunnel Vision): با پیشرفت بیماری، میدان دید به قدری کوچک میشود که گویی بیمار جهان را از درون یک لوله باریک نگاه میکند.
- افت دید مرکزی: در مراحل بسیار پیشرفته، درگیری ماکولا (مرکز شبکیه) باعث افت کیفیت دید حین مطالعه یا تشخیص چهرهها میشود.
روشهای تشخیص بیماری آرپی چشم؛ تست ERG و تصویربرداریهای پیشرفته
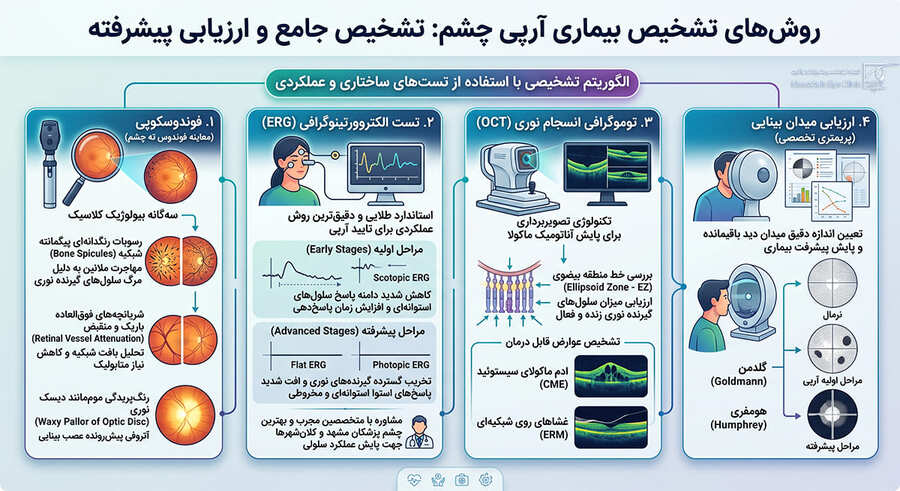
تشخیص قطعی و ارزیابی عوارض جانبی بیماری آرپی نیازمند استفاده ترکیبی از تستهای ساختاری و عملکردی الکتروفیزیولوژی است. یک پزشک متخصص فرآیند تشخیصی را از طریق الگوریتم زیر هدایت میکند:
۱. فوندوسکوپی (معاینه فوندوس ته چشم)
در طی معاینه بالینی شبکیه با افتالموسکوپ، سهگانه بیولوژیک کلاسیک بیماری آرپی خود را نشان میدهد:
- رسوبات رنگدانهای پیگمانته شبکیه به شکل اسپیکولهای استخوانی (Bone Spicules): ملانین لایه RPE به دنبال مرگ سلولهای گیرنده نوری مهاجرت کرده و به شکل رسوبات تیره سوزنی دور عروق شبکیه رسوب میکند.
- شریانچههای فوقالعاده باریک و منقبض شبکیه (Retinal Vessel Attenuation): به دلیل تحلیل رفتن بافت شبکیه و کاهش نیاز متابولیک به جریان خون رخ میدهد.
- رنگپریدگی دیسک نوری به صورت موممانند (Waxy Pallor of the Optic Disc): نشاندهنده شروع آتروفی پیشرونده عصب بینایی است.
۲. تست الکترورتینوگرافی (Electroretinography – ERG)
تست ERG استاندارد طلایی و دقیقترین روش عملکردی برای تایید بیماری آرپی چشم است. این آزمایش سیگنالهای الکتریکی تولیدشده توسط گیرندههای نوری شبکیه را در پاسخ به فلاشهای نوری ثبت میکند.
- در مراحل اولیه: دامنه (Amplitude) پاسخ سلولهای استوانهای (Scotopic ERG) بهشدت کاهش یافته و زمان پاسخدهی (Implicit Time) طولانی میشود. ارزیابیهای دقیق الکتروفیزیولوژیک از اهمیت بالایی برخوردار است و مراجعین محترم جهت پایش عملکرد سلولی شبکیه عموماً به دنبال مشورت با متخصصین مجرب و بهترین چشم پزشک مشهد یا سایر کلانشهرهای مجهز به این تجهیزات پاراکلینیکی میباشند تا سیر پاتولوژی را با دقت رصد نمایند.
- در مراحل پیشرفته: هر دو پاسخ استوانهای و مخروطی (Photopic ERG) بهشدت افت کرده و نمودار بهدستآمده کاملاً صاف (Flat ERG) میشود که به منزله تخریب گسترده گیرندههای نوری است.
۳. توموگرافی انسجام نوری (Optical Coherence Tomography – OCT)
تکنولوژی تصویربرداری OCT نقش بسیار مهمی در پایش آناتومیک ماکولا دارد. چشمپزشک با استفاده از OCT وضعیت خط منطقه بیضوی (Ellipsoid Zone – EZ) را بررسی میکند تا میزان سلولهای گیرنده نوری زنده و فعال باقیمانده را ارزیابی کند. همچنین عوارض شایع و قابل درمانی همچون ادم ماکولای سیستوئید (CME) یا تورم مرکز بینایی و غشاهای روی شبکیهای (ERM) با این ابزار تشخیص داده میشوند.
۴. ارزیابی میدان بینایی (Visual Field Testing / Perimetry)
پریمتری تخصصی (مانند گلدمن یا هومفری) اندازه دقیق میدان دید باقیمانده بیمار را مشخص کرده و پیشرفت بیماری را در طول زمان به صورت کمّی ثبت میکند.
درمان بیماری rp چشم
در حال حاضر (تا مقطع سال ۲۰۲۶)، درمان قطعی که بتواند تمام انواع آرپی را به طور کامل متوقف یا درمان کند وجود ندارد؛ اما علم با سرعت خیرهکنندهای در حال تغییر این واقعیت است.
ژندرمانی؛ چه کسانی کاندید دریافت “لوکستورنا” هستند؟
ژندرمانی مانند یک پیک دارویی عمل میکند؛ نسخه سالم ژن را مستقیماً به سلولهای بیمار میرساند. داروی لوکستورنا (Luxturna) نخستین درمان تایید شده توسط FDA است.
اما چه کسی آن را دریافت میکند؟ این دارو منحصراً برای بیمارانی است که اولاً جهش در ژن RPE65 دارند، و ثانیاً همچنان سلولهای زنده در شبکیهشان باقی مانده است. ژندرمانی نمیتواند سلول مرده را زنده کند، بلکه سلولهای در حال مرگ را نجات میدهد.
پروتزهای بینایی (چشم بیونیک)؛ تقابل فناوری پریما و اوریون
برای بیمارانی که در مراحل پیشرفته بیماری هستند و گیرندههای نوری خود را از دست دادهاند، پروتزها امیدبخش هستند.
- پروتز زیرشبکیهای (PRIMA): یک تراشه ۲ میلیمتری که در زیر شبکیه چشم کاشته میشود و با کمک عینک هوشمند، تصاویر را به پالسهای الکتریکی تبدیل میکند. این روش برای کسانی است که اعصاب چشمشان سالم است و میتواند امکان خواندن کلمات درشت را فراهم کند.
- پروتز قشری مغز (Orion): یک شاهکار مهندسی که به طور کامل چشم و عصب بینایی آسیبدیده را دور میزند و یک آرایه ۶۰ الکترودی مستقیماً روی قشر بینایی مغز کاشته میشود. این فناوری هنوز در مراحل کارآزمایی بالینی (مانند ارزیابیهای ۶ ساله EFS) قرار دارد و توانایی درک حرکت و نور (نقاط نوری یا فوسفن) را برای افراد کاملاً نابینا فراهم میکند.
سلولهای بنیادی؛ امیدهای آینده
درمان با سلولهای بنیادی با هدف جایگزینی سلولهای تخریبشده یا تزریق فاکتورهای محافظتکننده (نوروپروتکتیو) در حال انجام کارآزماییهای گسترده در آمریکا، روسیه و سایر کشورهاست. اگرچه نتایج اولیه درمانی امیدوارکننده است، اما هنوز به پروتکل استاندارد بالینی تبدیل نشده است.
جدول مقایسهای گزینههای درمانی آرپی چشم (آپدیت ۲۰۲۶)
| نوع تکنولوژی درمانی | نام روش / دارو | جامعه هدف و کاندیدای مناسب | اثربخشی و وضعیت بالینی |
| ژندرمانی ژن RPE65 | Luxturna | بیماران دارای جهش دوکپی در ژن RPE65 که هنوز سلولهای زنده در شبکیه دارند. | مورد تایید رسمی سازمانهای غذا و داروی جهانی (FDA و EMA)؛ بهبود جدی دید در تاریکی و حفظ بینایی. |
| ژندرمانی ژن RPGR | bota-vec | مردان مبتلا به فرم شدید وابسته به جنس (xlRP) ناشی از جهش RPGR. | نتایج درخشان کارآزمایی فاز ۳ (LUMEOS) در بهبود میدان بینایی؛ در مراحل نهایی فرآیند ثبت رگولاتوری. |
| سلولهای بنیادی (Stem Cells) | پیوند سلولهای مزانشیمی | بیماران در مرحله متوسط تا پیشرفته با هدف توقف مرگ سلولی. | فاز کارآزماییهای بالینی ۱ و ۲؛ بهبود ترشح فاکتورهای رشد برای نجات گیرندههای نوری رو به زوال. |
| تکنولوژی بیونیک زیرشبکیه | سیستم PRIMA | بیماران در فاز انتهایی آرپی که میدان دید مرکزی و بقیه گیرندههای نوری را از دست دادهاند. | تاییدیه ایمپلنتهای نسل جدید؛ کمک به بازگرداندن نسبی توانایی خواندن کلمات درشت و تشخیص اشیاء. |
| شبیهسازی مستقیم قشر مغز | سیستم کورتیکال Orion | بیماران دچار نابینایی مطلق ثانویه به آسیب شدید شبکیه یا تخریب عصب بینایی. | کارآزمایی بالینی بلندمدت فعال؛ ایجاد لکههای نوری در ذهن بیمار بدون استفاده از پاتووی چشم. |
تغذیه، مکملها و سبک زندگی؛ دروغها و واقعیتهای پزشکی
یکی از بزرگترین چالشهای بیماران، مواجهه با ادعاهای درمانی مکملهاست.
بررسیهای معتبر جهانی (مانند پایگاه علمی کاکرین – Cochrane) نشان میدهد که شواهد علمی برای اثربخشی مصرف دوز بالای ویتامین A یا روغن ماهی (DHA) در توقف آرپی بسیار ضعیف است. مصرف خودسرانه دوزهای بالای ویتامین A نه تنها به بینایی کمک چندانی نمیکند، بلکه میتواند به شدت برای کبد سمی باشد.
آنچه واقعاً موثر است: استفاده از عینکهای آفتابی جاذب اشعه ماوراء بنفش (UV) و فیلترهای نور آبی برای محافظت از سلولهای باقیمانده شبکیه.
به نقل از پایگاه داده مرورهای سیستماتیک کوکران (Cochrane)
متن انگلیسی:
“The evidence from randomized controlled trials regarding the efficacy of Vitamin A and fish oil (DHA) supplementation for delaying the progression of retinitis pigmentosa remains uncertain. Clinicians must weigh the potential minor benefits against the systemic risks of long-term high-dose vitamin A supplementation, especially in specific genetic subtypes.”
ترجمه فارسی:
«شواهد حاصل از کارآزماییهای بالینی تصادفیسازیشده در رابطه با اثربخشی مکملهای ویتامین A و روغن ماهی (DHA) در به تأخیر انداختن روند پیشرفت رتینیت پیگمنتوزا همچنان نامشخص است. پزشکان باید منافع بالقوه و اندک این مکملها را در برابر خطرات سیستمیک ناشی از مصرف طولانیمدت دوزهای بالای ویتامین A، بهویژه در زیرگروههای ژنتیکی خاص، بسنجند.»
زندگی با بیماری آرپی؛ توانبخشی بینایی و تجهیزات کمبینایان
مداخله توانبخشی کمبینایی (Low Vision Rehabilitation) حیاتیترین بخش مدیریت روزمره این بیماری است. استفاده از تلسکوپهای مینیاتوری، ذرهبینهای نوری، نرمافزارهای خوانش صفحه نمایش (Screen readers) و آموزشهای مسیریابی با عصای سفید، به بیماران کمک میکند تا با حفظ استقلال کامل به تحصیل، کار و زندگی اجتماعی خود ادامه دهند. در بسیاری از کشورها، نهادهای حمایتی نظیر “موسسه حمایت از بیماران چشمی آرپی” این خدمات و مشاورههای ژنتیک را ارائه میدهند.
نکته حرفهای و تجربهمحور (Clinical Pearl): مدیریت عوارض جانبی
پزشکان باتجربه میدانند که آرپی به تنهایی عمل نمیکند. بسیاری از بیماران مبتلا به آرپی، در سنین پایینتر دچار «آب مروارید» (Cataract) و یا «ورم مرکز شبکیه» (CME) میشوند. افت ناگهانی دید در یک بیمار آرپی ممکن است ناشی از پیشرفت خود بیماری نباشد، بلکه به دلیل ورم شبکیه باشد که با قطرهها یا داروهای خوراکی به راحتی قابل درمان و بازگشت است. همواره پیگیریهای دورهای منظم با بهترین چشم پزشک مشهد یا پزشک معتمد در شهر محل سکونتتان ضروری است تا این عوارض ثانویه به سرعت تشخیص و درمان شوند.
⚖️ هشدار مهم (Important Warning): تعریف نابینایی قانونی
زمانی که میدان دید بیمار به کمتر از ۲۰ درجه برسد (ایجاد دید تونلی شدید)، فرد از نظر حقوقی و پزشکی کاندید «نابینایی قانونی» (Legal Blindness) محسوب میشود؛ حتی اگر دید مرکزی او تیز و شفاف باشد. در این شرایط، به دلیل خطرات جدی، رانندگی اکیداً ممنوع بوده و بیمار برای حفظ ایمنی خود نیازمند استفاده از تجهیزات مسیریابی است.
آیا بیماری آرپی چشم باعث نابینایی کامل میشود؟ (پیشآگهی بالینی)
این دغدغه ذهنی، شایعترین علت مراجعه و اضطراب بیماران آرپی و خانوادههای آنان است. از منظر کلینیکی، روند بیماری گرچه پیشرونده است اما با نابینایی مطلق تفاوتهای زیادی دارد:
- حفظ دید مرکزی تا فاز انتهایی: در اکثریت قاطع بیماران، به دلیل بقای سلولهای مخروطی در ناحیه فووهآ (مرکز ماکولا)، تیزبینی مرکزی و توانایی خواندن تا سنین میانسالی یا کهنسالی حفظ میشود. بنابراین، بیمار گرچه میدان دید خود را از دست میدهد (دید لولهای)، اما کاملاً تاریک و نابینا نمیشود.
- نابینایی قانونی (Legal Blindness): طبق تعاریف پزشکی، اگر میدان دید بیمار به کمتر از ۲۰ درجه کاهش یابد، حتی در صورت خوب بودن دید مرکزی، بیمار از نظر قانونی کمبینای شدید یا نابینای قانونی تلقی میشود و از رانندگی و کارهای نیازمند دید پیرامونی وسیع محروم میگردد.
- نقش سن شروع علائم: هرچه علائم بالینی در سنین پایینتر (کودکی) بروز یابد، سرعت پیشرفت بیماری بیشتر بوده و پیشآگهی بینایی ضعیفتر است. در مقابل، شروع علائم در بزرگسالی با پیشرفت بسیار ملایمتری همراه است.
جدول شماره ۱: مقایسه تستهای تشخیصی ضروری در آرپی
| نوع آزمایش | هدف اصلی بالینی | کاربرد ویژه در بیماران آرپی |
| OCT (توموگرافی شبکیه) | عکسبرداری سهبعدی از لایههای شبکیه | پایش میزان ضخامت شبکیه و تشخیص زودهنگام ورم ماکولا (CME) |
| ERG (الکترورتینوگرام) | سنجش فعالیت الکتریکی سلولهای چشم | تایید قطعی بیماری؛ در آرپی پاسخ الکتریکی سلولها خاموش یا بسیار ضعیف است |
| آزمایش ژنتیک | نقشهبرداری DNA و یافتن ژن جهشیافته | تعیین احتمال انتقال به فرزندان و مشخص کردن کاندیداتوری برای ژندرمانی |
جدول شماره ۲: مقایسه کاربردی فناوریهای پیشرفته درمانی
| فناوری / روش | بیماران کاندید (چه کسانی سود میبرند؟) | مکانیسم اثر | سطح انتظار واقعبینانه |
| ژندرمانی (Luxturna) | بیماران دارای جهش RPE65 با سلولهای زنده | توقف پیشرفت بیماری و حفظ دید موجود | بازگشت دید طبیعی رخ نمیدهد؛ صرفاً جلوگیری از بدتر شدن. |
| پروتز زیرشبکیه (PRIMA) | بیماران با تخریب گیرندههای نوری اما عصب سالم | تحریک سلولهای عصبی باقیمانده چشم | دیدن نقاط نورانی و توانایی خواندن حروف بسیار درشت. |
| پروتز قشری مغز (Orion) | بیماران با نابینایی مطلق و عصب چشم کاملاً تخریبشده | دور زدن کامل چشم و تحریک مستقیم قشر مغز | تشخیص جهت حرکت و منبع نور (در حد جهتیابی پایهای). |
🔬 جمعبندی بیماری RP یا رتینیت پیگمانتوزا را چقدر می شناسید؟
علم چشمپزشکی در مواجهه با بیماری رتینیت پیگمنتوزا در یک دوران گذار تاریخی قرار دارد. تا یک دهه پیش، آرپی مترادف با یک “حکم قطعی کاهش بینایی بدون هیچ راهحلی” بود. امروزه، با ورود فناوری کریسپر (CRISPR)، ژندرمانیهای هدفمند و توسعه رابطهای مغز-رایانه (نظیر ایمپلنتهای Orion)، ادبیات بالینی از واژه «غیرقابل درمان» به سمت «بیماری نیازمند مدیریت ژنتیکی و توانبخشی» تغییر جهت داده است.
مهمترین توصیه تخصصی به همکاران بالینی و بیماران این است: زمان طلایی را از دست ندهید. تعیین دقیق نقشه ژنتیکی بیمار، ارجاع زودهنگام به مراکز توانبخشی کمبینایی، و پایش مستمر عوارض ثانویه (مانند کاتاراکت)، مؤثرترین رویکرد مبتنی بر شواهد (EBM) برای حفظ بالاترین سطح از کیفیت زندگی این بیماران تا رسیدن به تجاریسازی کامل درمانهای نوین است.